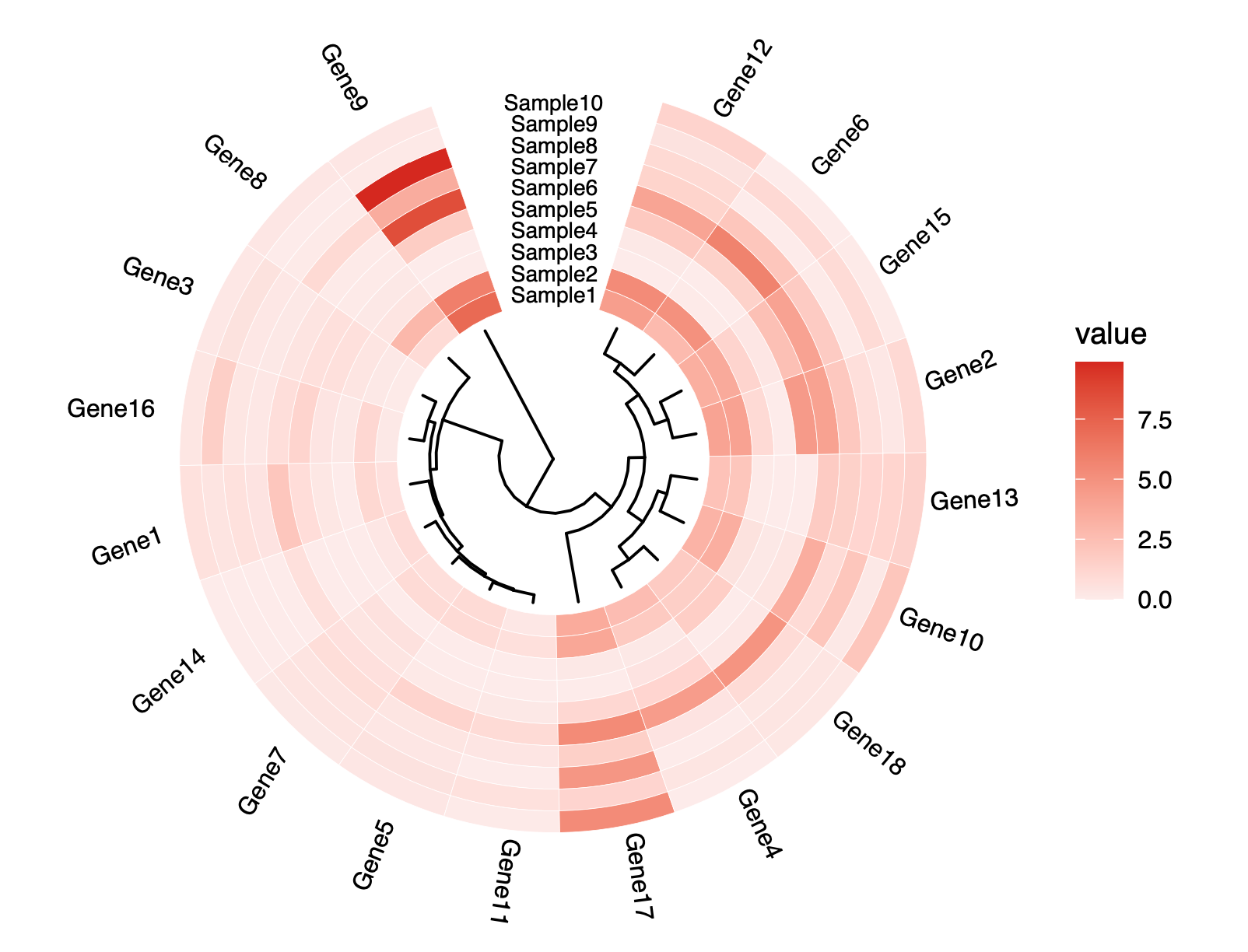
基因功能富集分析

基因功能富集分析
泡泡基因功能富集分析算法
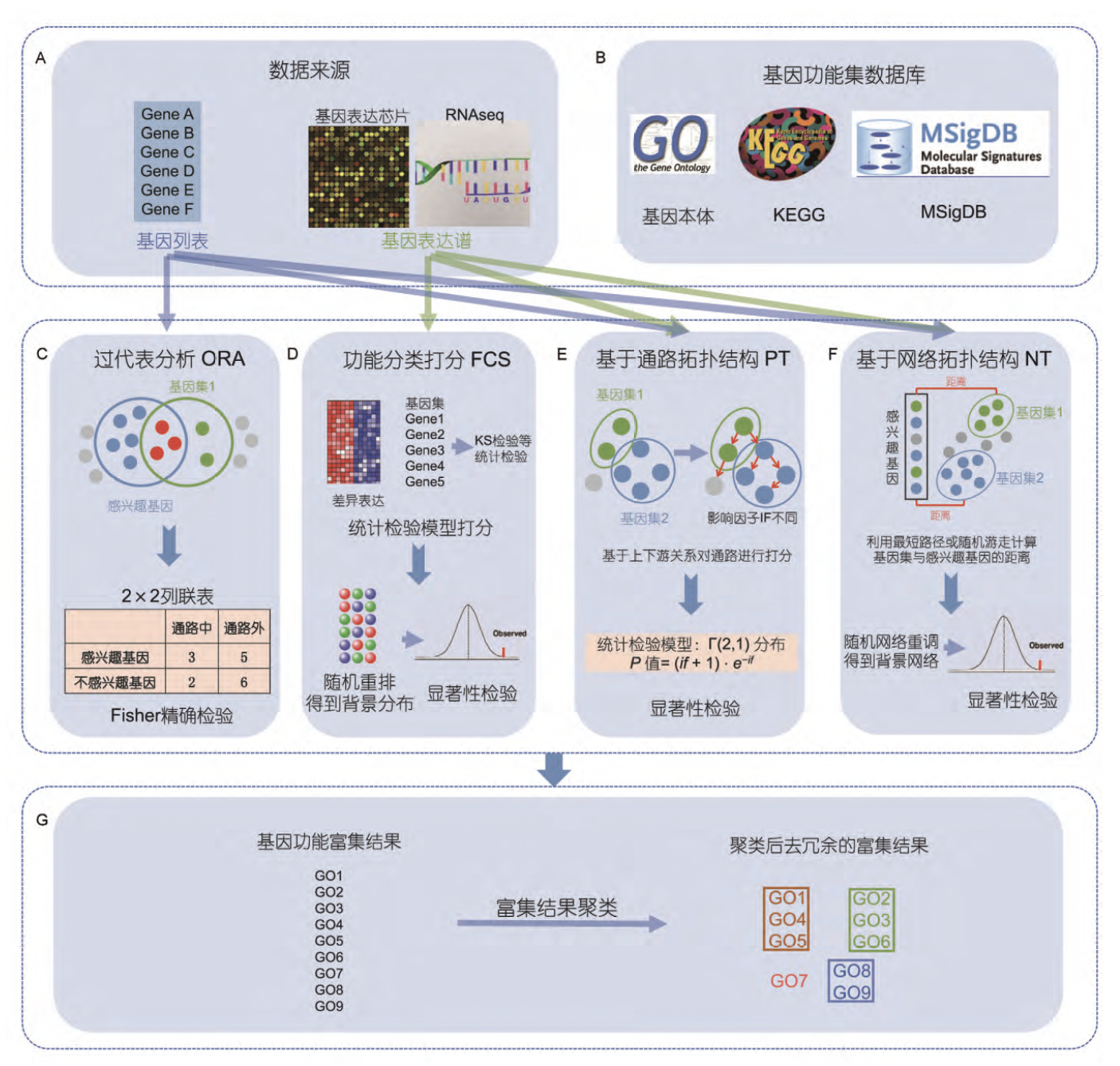
over-representation analysis: 先计数基因列表与基因功能集共同的基因,并利用2×2列联表,进行Fisher精确检验。根据超几何分布来检验基因列表中的基因在待测功能集中是否显著富集。
优点:
- 比较完备的统计学理论,输出结果比较稳健、可靠
缺点:
- 它仅仅使用了基因的数目信息,而没有利用基因表达水平或表达差异值
- 通常仅使用最显著的基因序列,而忽略差异不显著的基因, 导致检测灵敏性的降低
- 将每一个基因进行同等对待,它忽视了基因在通路内部生物学意义的不同,以及基因间复杂的相互作用
- 假设通路与通路间是独立的,但这个前提假设是不太准确的
GO/KEGG统计学方法详解
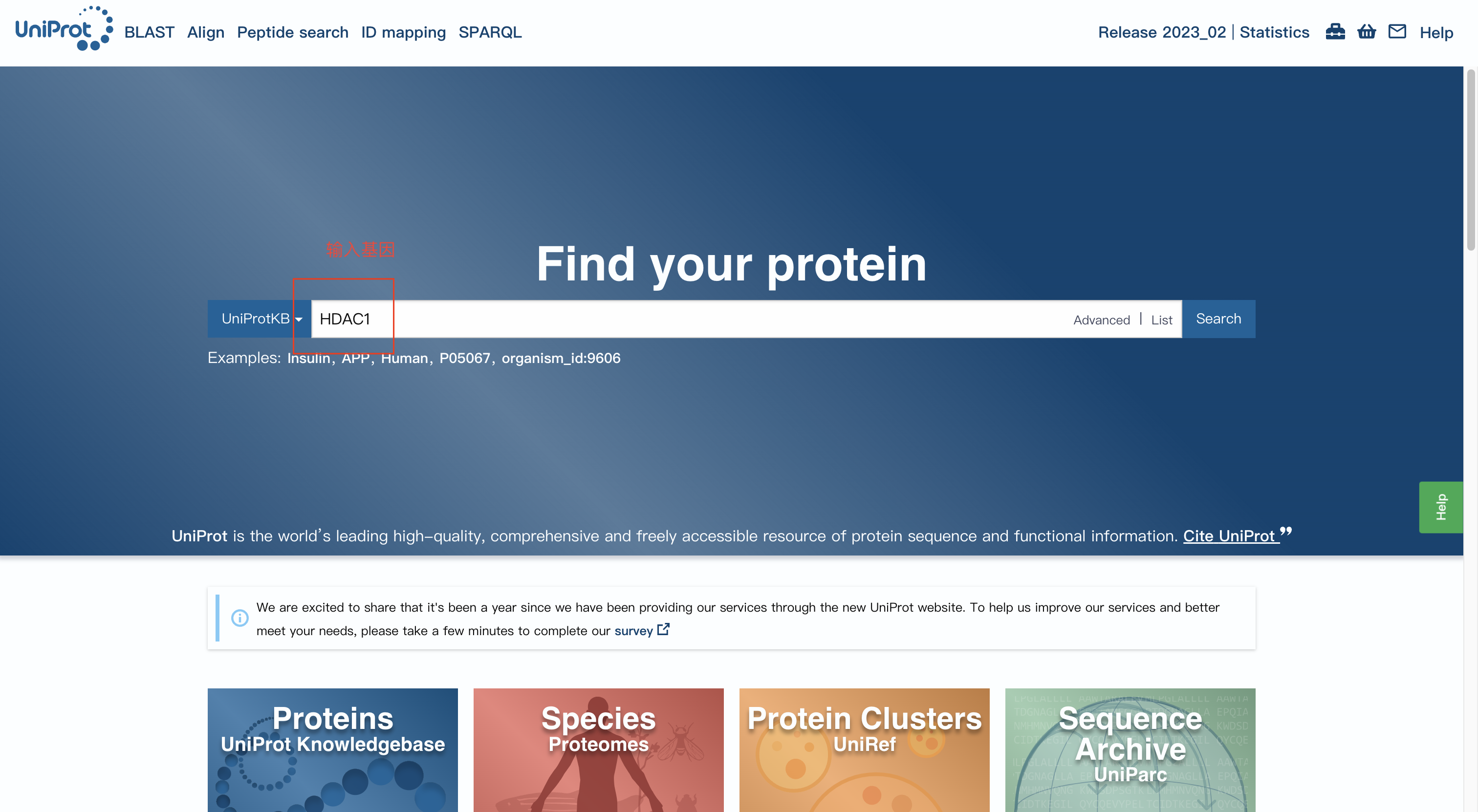
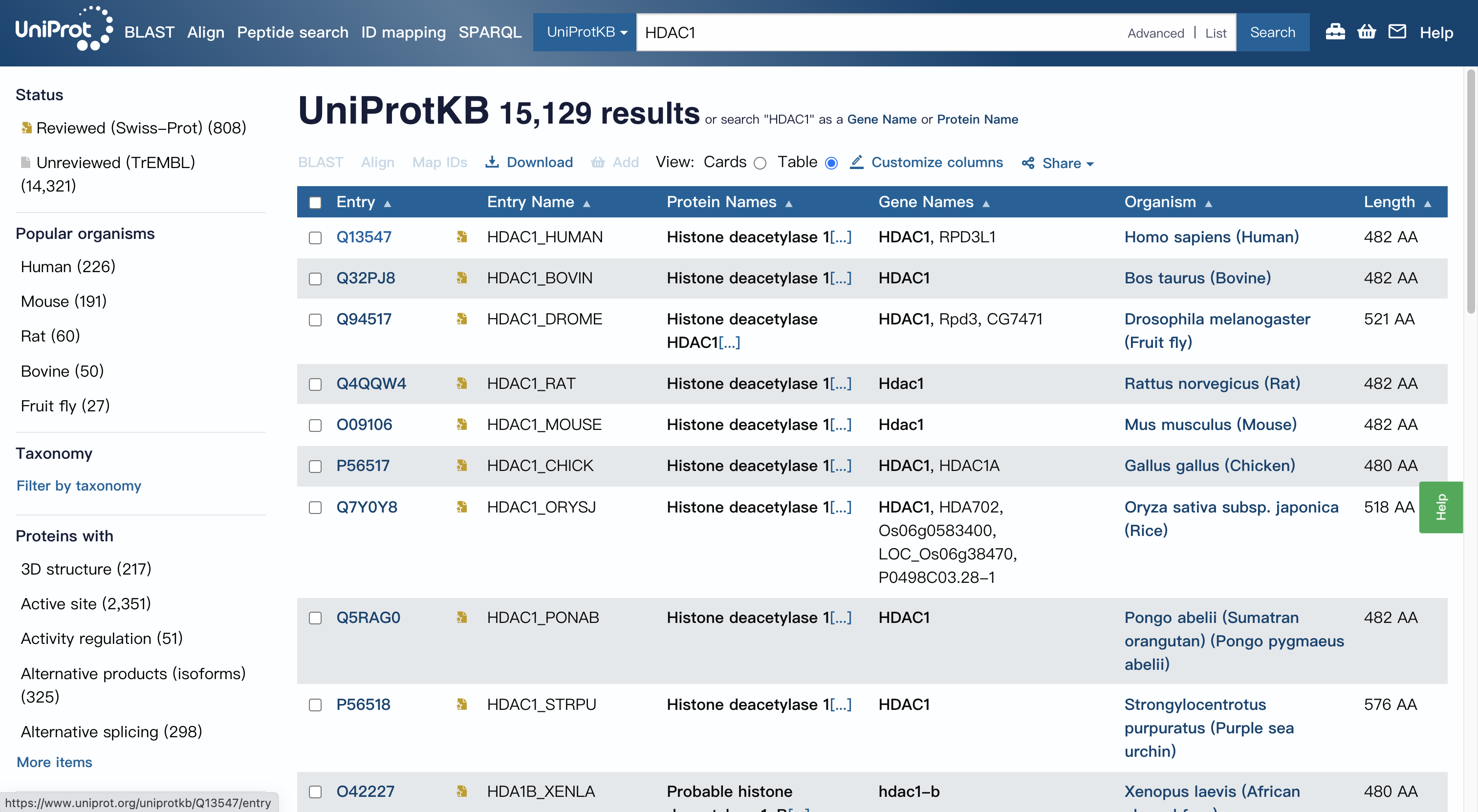
目前有很多工具和数据库提供ORA的使用,常见的就是GO富集分析和KEGG富集分析。我们通常通过处理二代测序和芯片测序得到的数据,进而设置一定对阈值,如差异倍数或P值,得到的一组我们感兴趣的基因,进而导入这个基因的ID,对这些基因进行注释。
GO富集分析将导入的每一个基因分别对应的CC、BP、MM进行罗列,使用超几何分布来确定这些对应的通路是否是显著性富集:
N: GO注释数据库中的总基因数
M:数据库中属于某个GO子类的基因数
n: 进行GO富集分析的基因总数目
k: n中属于M的数目KEGG富集分析结果结果体现在由于处理导致个体或组基因表达发生较大改变,导致通路有较大改变。原理和上面类似。
functional class scoring: 以GSEA为例, 对基因表达谱中的所有基因按照与表型的差异表达程度排序,再利用KS检验对待测功能基因集打分,利用随机重排得到背景分布,找出显著富集的功能基因集。
优点:
- 考虑到了基因表达值的属性信息问题
- 以待测基因功能集为对象来进行检验,使得检测结果更加灵敏及更有针对性
缺点:
- 独立分析每一条通路,但同一个基因可能涉及多条通路,不同通路间的基因出现重叠,所以可能会导致别的通路由于重叠基因问题,也出现显著富集现象。
- 仍把待测基因功能集中的每个基因作为独立的个体, 忽略了基因的生物学属性和基因间的复杂相互作用关系。
GSEA统计学方法详解
GSEA是常用的一种FCS方法. 其基本思路是首先基于表达差异值对全基因组基因进行排序得到基因列表, 然后检验待测基因功能集中的基因相对于随机情况而言, 是否显著地位于基因列表的顶端或底端, 即待测基因集的表达水平在案例和对照实验状态下是否发生了明显的变化.具体而言, GSEA 首先计算了每个基因的表达水平与案例和对照两种状态下的关联系数,并对关联系数从高到低进行了排序; 然后, 针对一特定的待测基因功能集, 根据其中每个基因的排序情况,利用加权的近似KS检验, 获得待测基因功能集在排序列表中的KS检验值————也即待测基因功能集的统计值; 为检验观察统计值的显著性, GSEA 通过对样本的随机排列来获得统计值的背景分布, 并利用该分布来评估观察统计值的P值.除对样本的随机排列外, 在样本量较少的情况下, GSEA也可用对基因的随机排列来估算待测基因功能集的显著水平。

pathway topology: 以Pathway-Express为例,根据上下游关系对通路进行打分,上游通路影响因子较大,对待测基因功能集根据影响因子进行打分,然后根据统计检验模型计算显著程度。
优点:
- 基于PT的基因功能富集算法对研究一些比较完善、拓扑结构完整的通路会有更强的显著性。
缺点:
- 研究较少,信息不完善的通路稳健型较差
network topology: 以NEA为例,将基因列表或基因表达谱和待测基因功能集放入生物网络中,计算感兴趣的基因与待测基因功能集在网络之间的最短距离,通过对网络重调得到背景分布,找出显著富集的基因功能集。
优点:
- 加入了系统层面的基因重要性程度及关联信息等属性数据,使得预测结果更加准确可靠
缺点:
- 更多信息的加入也容易导致算法过于复杂,计算速度较慢,实现难度也就更大。
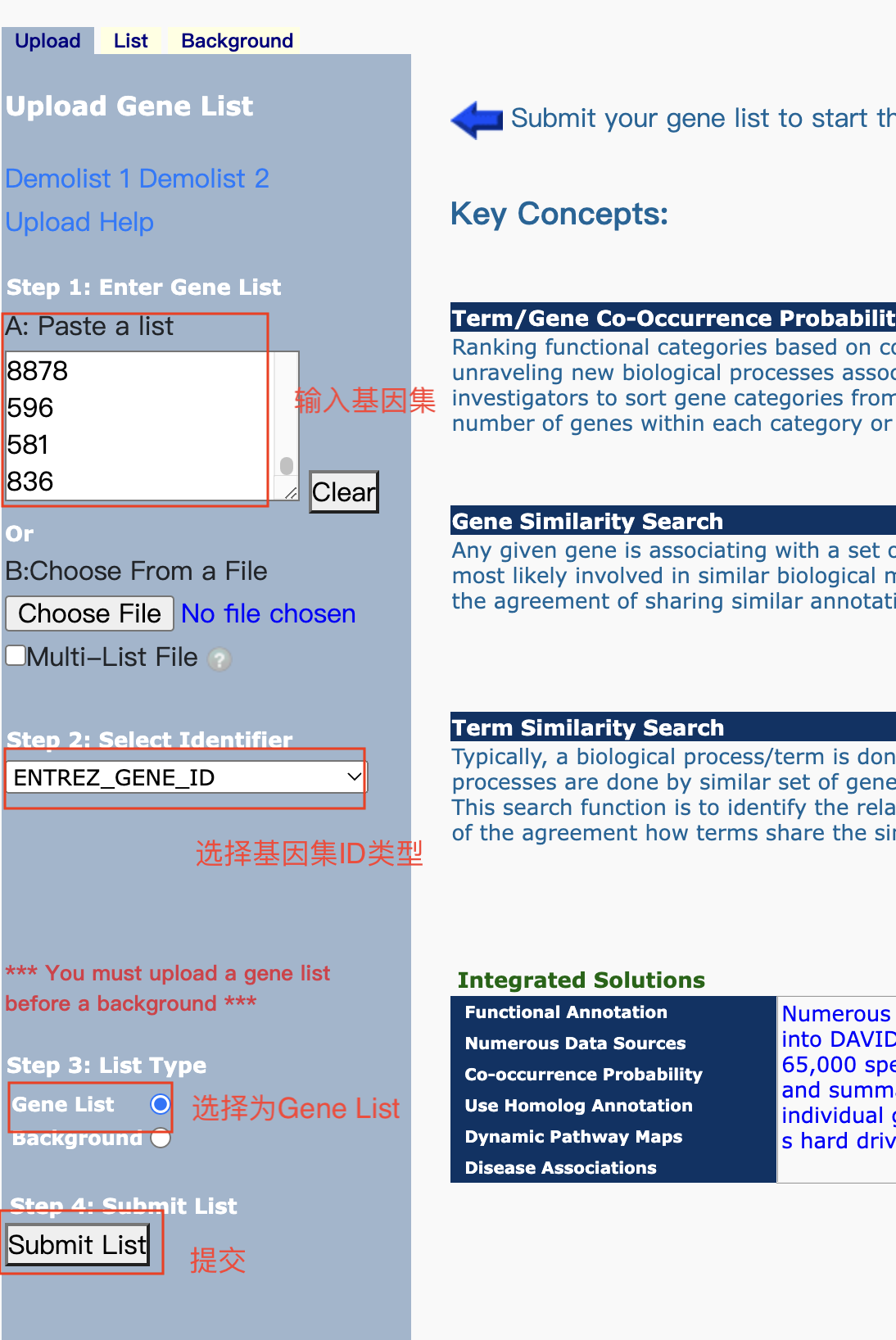
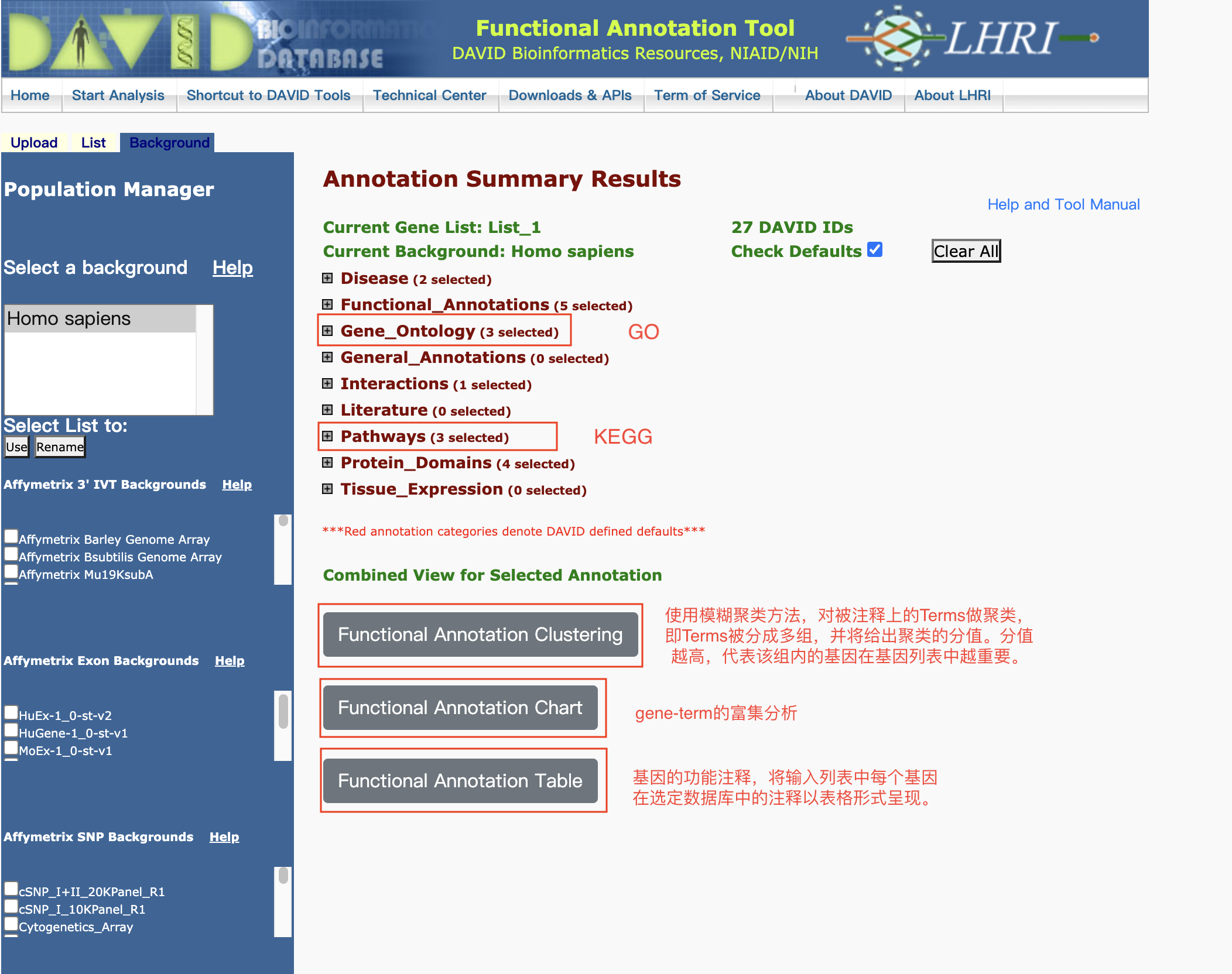
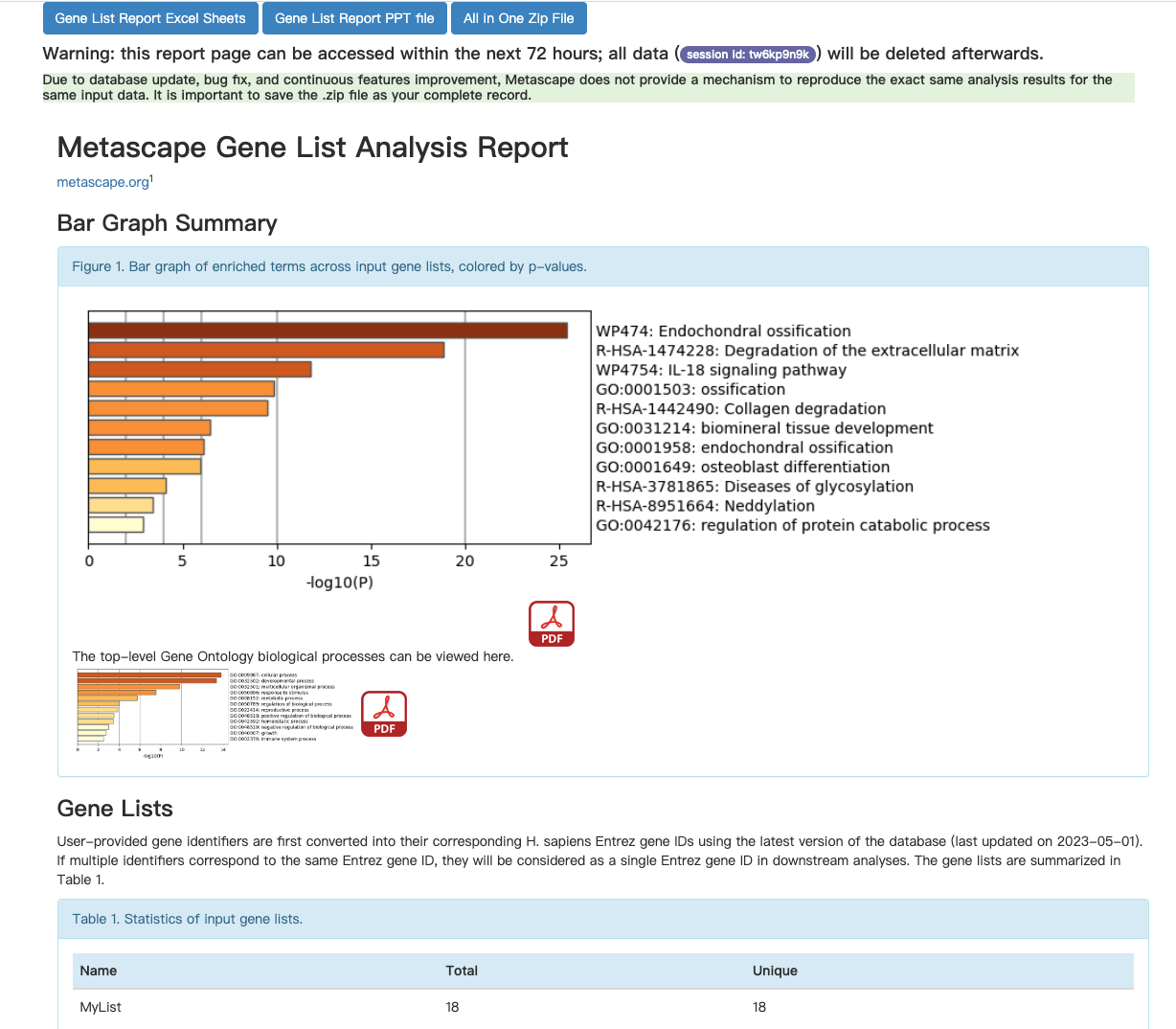
富集分析在线工具
- 基因/蛋白功能注释(注释模块)和功能基因集富集(富集模块)的在线工具
- 注释模块:输入基因列表,从通路、疾病、基因本体论等多个数据库中提取基因功能注释信息
- 富集模块:输入基因列表或基因表达数据,根据多种方法生成富集的基因集、对应的名称、P值或富集概率和富集分数
- 支持microarray和RNA-Seq数据的分析
- 功能富集分析在线工具
- 支持三种富集分析方法:ORA、GSEA、NTA
- 综合性基因富集分析在线工具
- 支持BED格式和基因列表的输入
- 富集分析的数据库包括Transcription(转录), Pathways(通路), Ontologies(GO), Diseases/Drugs(疾病/药物), Cell types(细胞类型), Misc(未分类数据集), Legacy, Crowd
- miRNA富集网站
- 将miRNA分为六大类:miRNA-family sets(miRNA家族),miRNA cluster sets(miRNA聚类),miRNA-disease(miRNA疾病),miRNA-function sets(miRNA功能),miRNA-TF sets(miRNA与转录因子),tissue specificity sets(组织特异性)。
- “Analysis”:对一个miRNA列表,以数据库miRNA集为背景进行富集分析
- “Comparison”:对一组上调和下调的miRNA列表,评估上下调的miRNA与疾病发生或抑制的关系
- “Query”:通过关键词查询相关的miRNA集
- 单细胞分析在线工具
GO/KEGG富集分析
Gene Ontology: GO的初衷就是希望提供一个能对基因和基因产物特性相关的术语或词义 进行描绘及解释的工作平台,从而使生物信息学研究者对基因和基因产物的数据能够进行统一的归纳、处理、解释和共享,便于我们理解和沟通。 基因本体涉及的基因和基因产物词汇分为三大类,涵盖生物学的三个方面。
GO term 分类
细胞组分(cellular component):细胞的每个部分和细胞外环境。在哪里发挥作用:往往用于描述基因在细胞上的定位信息,当研究者关注一些特殊的亚细胞定位研究的时候具有重要的意义。
分子功能(molecular function):可以描述为分子水平的活性(activity),如催化(catalytic)或结合(binding)活性等。做什么工作:用于描述蛋白的实际作用方式,当研究者更关注整个生物学事件中蛋白作用方式上的变化时,可以考虑从这个角度切入。
生物过程(biological process):是指由一个或多个分子功能有序组合而产生的系列事件,一般一个过程是由多个不同的步骤组成。目的是什么:更贴近表型,往往可以很好地描述样本的实际情况。
通路数据库 KEGG


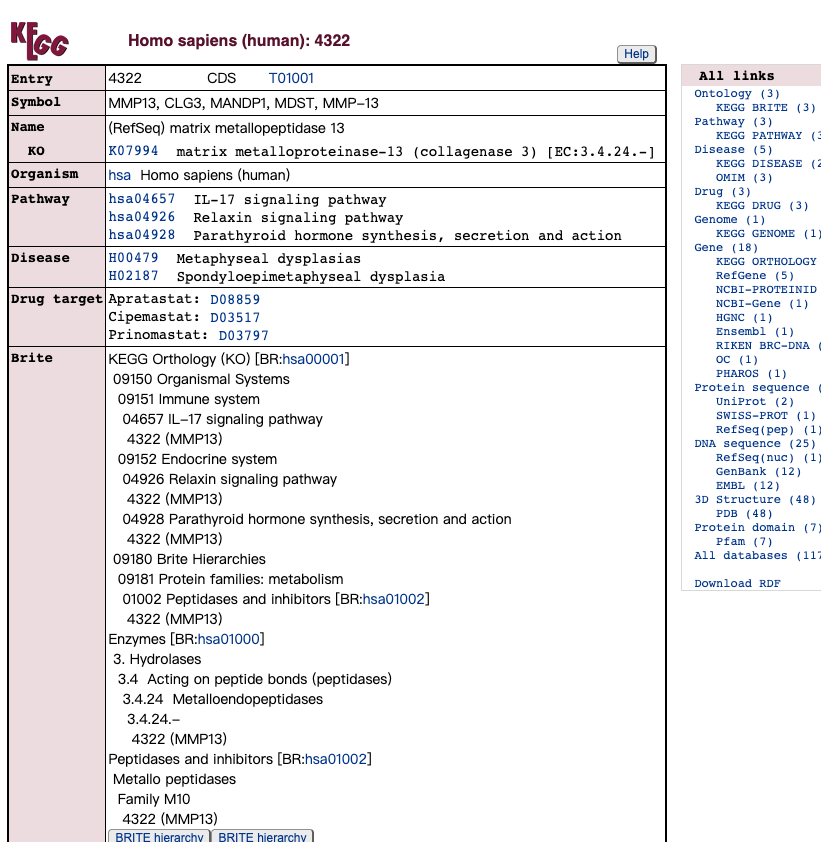
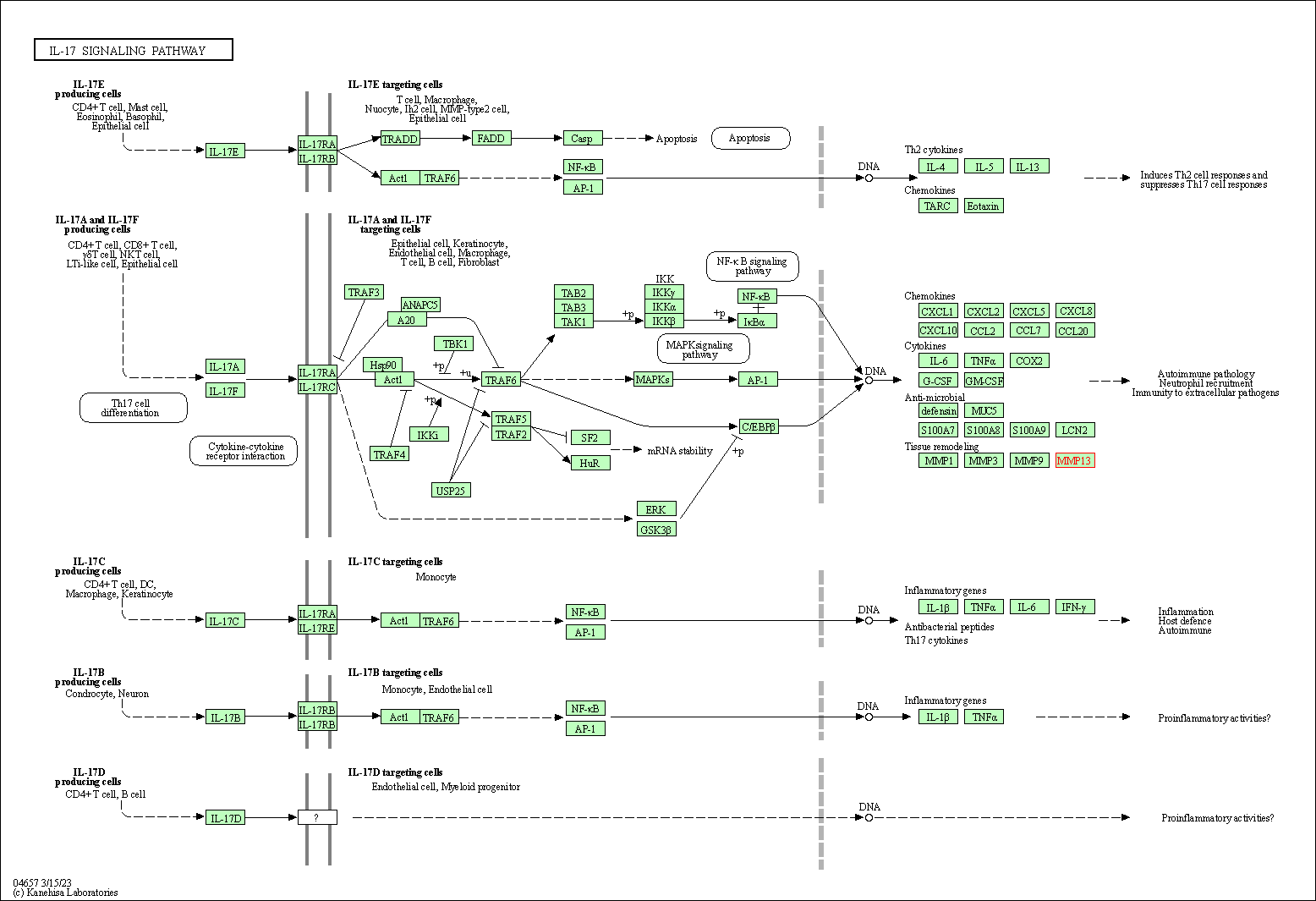

通路数据库 Reactome
- 通路知识的可视化、解释和分析提供直观的生物信息学工具。
- Reactome是reaction(反应)+ ome(组)的合成词,反应可以被视为途径的“步骤”。Reactome将“反应”定义为生物学中任何改变生物分子状态的事件, 涉及催化剂的结合、活化、易位、降解和经典生物化学事件。因此,Reactome数据模型的核心单元是反应(reaction),参与反应的实体(核酸、蛋白质、复合物剂和小分子)形成生物相互作用的网络并被分组成通路。
1 进入官网,选择Pathway Browser
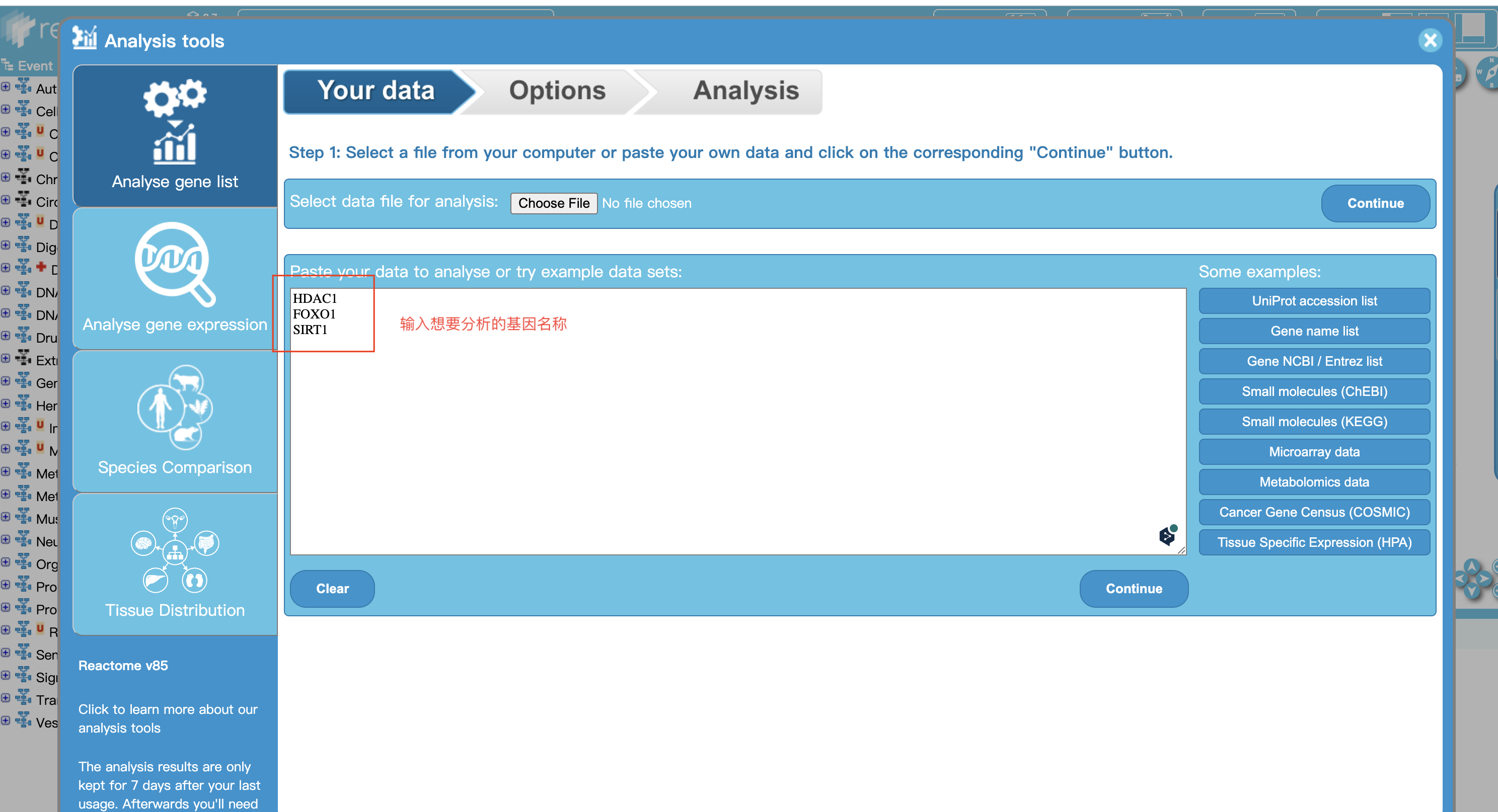
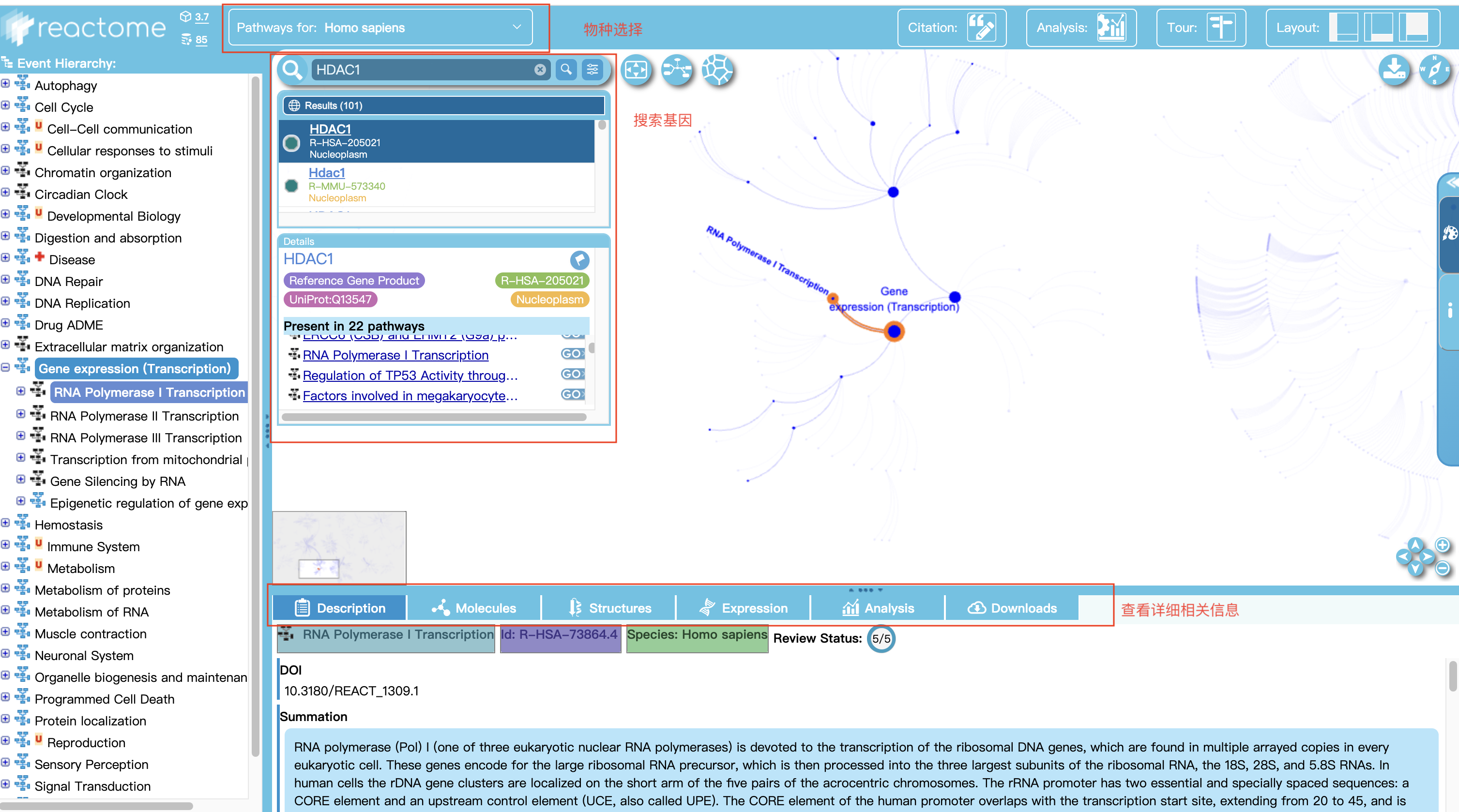
2 输入相关信息查看结果
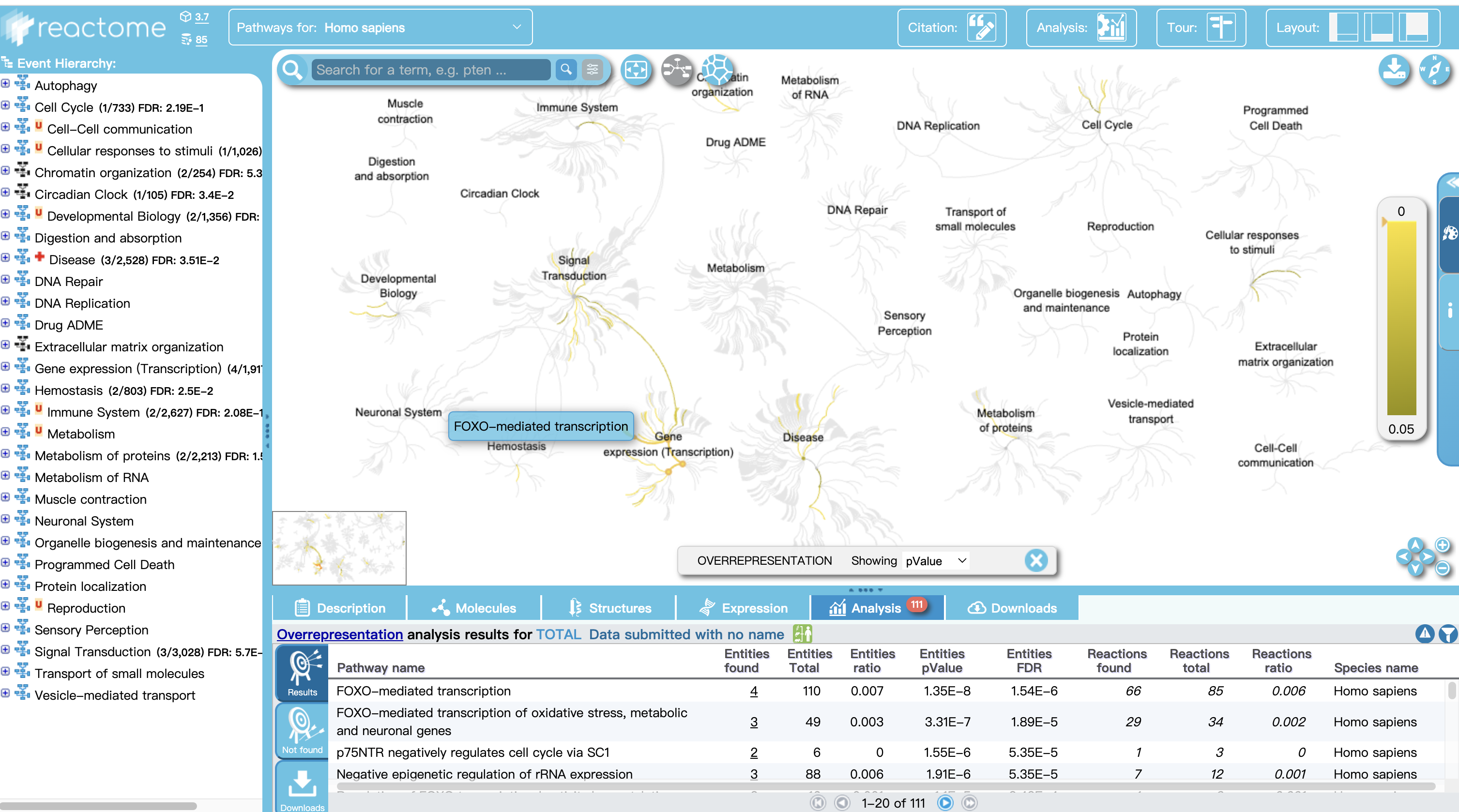
3 也可进入Analysis Tools进行多个基因通路查看