第 11 章 TCGA 生存分析
11.0.1 临床信息下载
查看 TGGA 项目
# 加载包
library(TCGAbiolinks)
library(stringr)
library(SummarizedExperiment)
getGDCprojects()$project_id %>% str_subset("TCGA")
proj = "TCGA-PAAD"下载临床数据
f2 = paste0(proj,"clf.Rdata")
if(!file.exists(f2)){
query = GDCquery(project = proj,
data.category = "Clinical",
data.type = "Clinical Supplement",
data.format = "BCR XML"
)
GDCdownload(query)
dat = GDCprepare_clinic(query, clinical.info = "patient")
k = apply(dat, 2, function(x){!all(is.na(x))});table(k)
clinical = dat[,k]
save(clinical,file = f2)
}
load(f2)11.0.2 生存信息下载
xena 单独整理了生存信息,可以使用它的数据 https://shixiangwang.github.io/home/en/tools/ucscxenatools-intro/
library(UCSCXenaTools)
library(dplyr)
cohort <- XenaData %>%
filter(XenaHostNames == "tcgaHub") %>% # select TCGA Hub
XenaScan("PAAD")
cli_query <- cohort %>%
filter(DataSubtype == "phenotype") %>% # select clinical dataset
XenaGenerate() %>% # generate a XenaHub object
XenaQuery() %>%
XenaDownload()
cli <- XenaPrepare(cli_query)
surv <- cli[[2]]11.1 合并生存信息和临床信息
meta <- left_join(surv, clinical, by = c("_PATIENT"= "bcr_patient_barcode"))
nrow(meta)
length(unique(meta$sample))
meta <- distinct(meta, sample, .keep_all = T)11.1.0.2 规范列名
选择需要的列,简化列名
tmp <- data.frame(colnames(meta))
meta <- meta[,c(
'sample', # 样本 ID
'OS', # 结局事件
'OS.time', # 从生存期
'race_list', # 种族
'age_at_initial_pathologic_diagnosis', # 年龄
'gender' , # 性别
'stage_event_pathologic_stage' # 病理分期
)]
colnames(meta)=c('ID','event','time','race','age','gender','stage')
str(meta)
meta$gender = as.character(meta$gender)
meta$stage = as.character(meta$stage)
meta$race = as.character(meta$race)11.2 表达数据下载
下载表达矩阵
proj = "TCGA-PAAD"
f1 = paste0(proj, "expf.Rdata")
if(!file.exists(f1)){
query = GDCquery(project = proj,
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "STAR - Counts",
)
GDCdownload(query)
dat = GDCprepare(query) # 创建 SummarizedExperiment 对象
exp = assay(dat) # 提取表达矩阵
tpm = assay(dat,4) # 提取 TPM 矩阵
save(exp, tpm, file = f1)
}
load(f1)
# 若生存分析使用 TPM (可选)
exp <- tpm11.2.2 去除 normal 样本
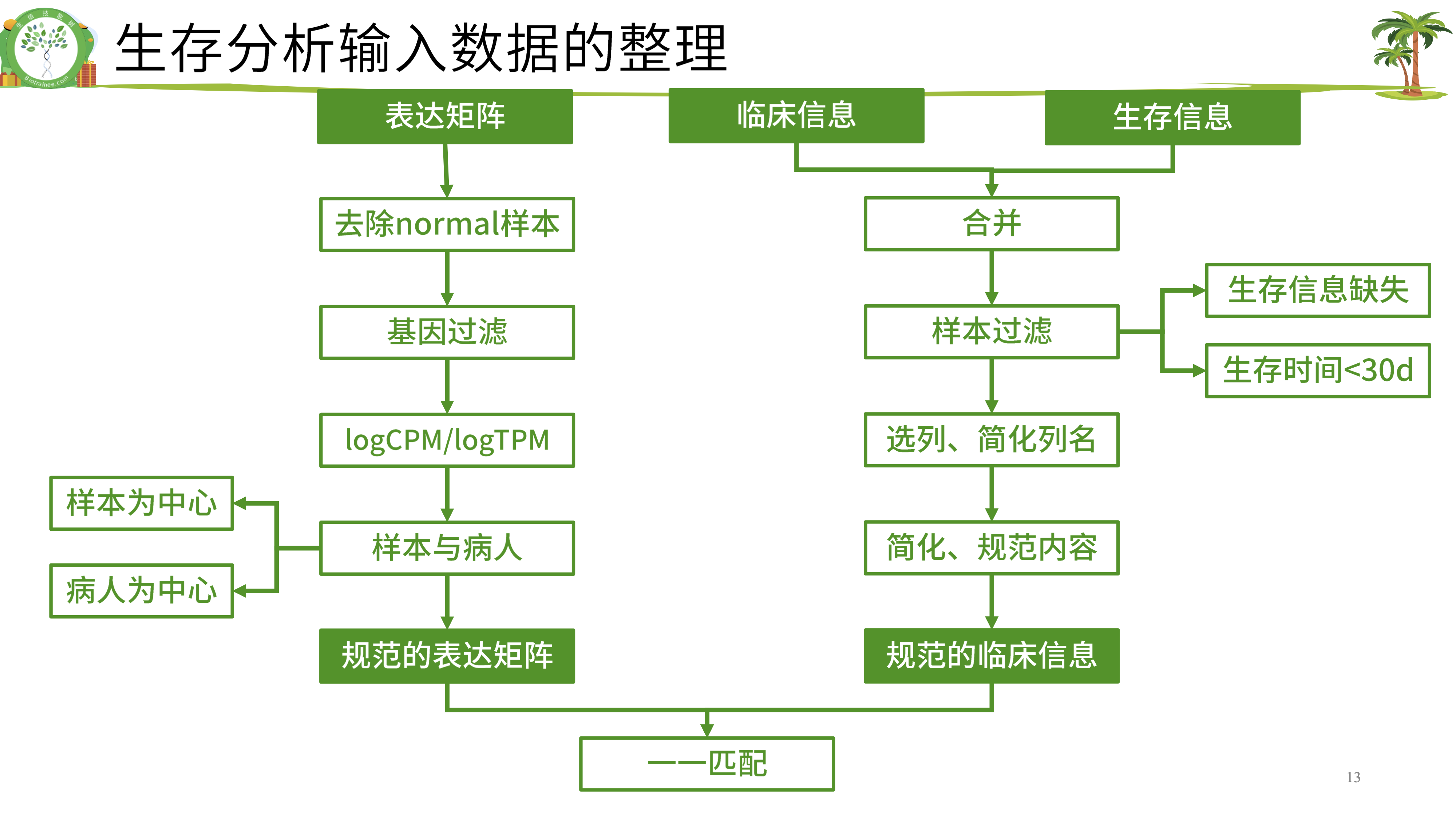
表达矩阵只需要tumor数据,不要normal,将其去掉,新表达矩阵数据命名为 exprSet。 临床信息需要进一步整理,成为生存分析需要的格式,新临床信息数据命名为 meta。 由于不同癌症的临床信息表格列名可能不同,这里的代码需要根据实际情况修改。
分组信息获取
11.2.3 基因过滤
(1)标准1:至少要在50%的样本里表达量大于0(最低标准)。 exp(600样本)满足“至少在300个样本里表达量>0”,不能等同于 exprSet(500样本)满足“至少在250个样本里表达量>0”。
k = apply(exprSet,1, function(x){sum(x>0)>0.5*ncol(exprSet)});table(k)
exprSet = exprSet[k,]
nrow(exprSet)(2)标准2:至少在一半以上样本里表达量>10(其他数字也可,酌情调整)
11.3 表达矩阵与临床信息匹配
即:meta的每一行与exprSet每一列一一对应
rownames(meta) <- meta$ID
head(rownames(meta))
head(colnames(exprSet))
colnames(exprSet) <- str_sub(colnames(exprSet),1,15)
s <- intersect(rownames(meta),colnames(exprSet));length(s)
exprSet <- exprSet[,s]
meta <- meta[s,]
rownames(meta) <- meta$ID
dim(exprSet)
dim(meta)
identical(rownames(meta), colnames(exprSet))
save(meta, exprSet, proj, file = paste0("Rdata/", proj,"_sur_model.Rdata"))11.4 生存分析
rm(list = ls())
proj = "TCGA-PAAD"
load("Rdata/", paste0(proj,"_sur_model.Rdata"))
exprSet[1:4,1:4]
str(meta)11.4.2 连续型变量
11.4.2.1 年龄
group <- ifelse(meta$age > median(meta$age, na.rm = T),"older","younger")
table(group)
sfit=survfit(Surv(time, event)~group, data=meta)
ggsurvplot(sfit,pval =TRUE, data = meta, risk.table = TRUE)#最佳截断值-年龄
# 通过遍历所有可能的分割点,计算每个分割点对应的统计量(如 log-rank 统计量),然后选择使统计量最大的分割点。
cut <- surv_cutpoint(meta, time = "time", event = "event",
variables = "age")
m <- cut[["cutpoint"]][1, 1]
group <- ifelse(meta$age > m,"older","younger")
table(group)
sfit <- survfit(Surv(time, event)~group, data=meta)
ggsurvplot(sfit,pval =TRUE, data = meta, risk.table = TRUE)11.4.2.2 基因
# 中位数截断
g <- rownames(exprSet)[1];g
meta$gene <- ifelse(exprSet[g,] > median(exprSet[g,]),'high','low')
sfit <- survfit(Surv(time, event)~gene, data=meta)
ggsurvplot(
sfit,
data = meta,
pval = TRUE,
risk.table = TRUE,
title = paste("Survival Curve for", g),
legend.labs = c("High Expression", "Low Expression")
)#最佳截断值-基因
dat <- meta
dat$gene2 = exprSet[g,]
cut = surv_cutpoint(dat, time = "time", event = "event",
variables = "gene2")
m = cut[["cutpoint"]][1, 1]
dat$gene = ifelse(exprSet[g,]>m,'high','low')
table(dat$gene)
sfit=survfit(Surv(time, event)~gene, data=dat)
ggsurvplot(sfit,pval =TRUE, data = dat, risk.table = TRUE)11.4.3 log-rank test
KM的p值是log-rank test得出的,可以批量操作
# KM_cox_function.R
geneKM <- function(gene){
meta$group=ifelse(gene>median(gene),'high','low')
data.survdiff=survdiff(Surv(time, event)~group,data=meta)
p.val = 1 - pchisq(data.survdiff$chisq, length(data.survdiff$n) - 1)
return(p.val)
}
genecox <- function(gene){
meta$gene = gene
#可直接使用连续型变量
m = coxph(Surv(time, event) ~ gene, data = meta)
#也可使用二分类变量
#meta$group=ifelse(gene>median(gene),'high','low')
#meta$group = factor(meta$group,levels = c("low","high"))
#m=coxph(Surv(time, event) ~ group, data = meta)
beta <- coef(m)
se <- sqrt(diag(vcov(m)))
HR <- exp(beta)
HRse <- HR * se
#summary(m)
tmp <- round(cbind(coef = beta,
se = se, z = beta/se,
p = 1 - pchisq((beta/se)^2, 1),
HR = HR, HRse = HRse,
HRz = (HR - 1) / HRse,
HRp = 1 - pchisq(((HR - 1)/HRse)^2, 1),
HRCILL = exp(beta - qnorm(.975, 0, 1) * se),
HRCIUL = exp(beta + qnorm(.975, 0, 1) * se)), 3)
return(tmp['gene',])
#return(tmp['grouphigh',])#二分类变量
}# source("KM_cox_function.R")
logrankfile <- paste0(proj,"_log_rank_p.Rdata")
if(!file.exists(logrankfile)){
log_rank_p <- apply(exprSet , 1 , geneKM)
log_rank_p <- sort(log_rank_p)
save(log_rank_p, file = logrankfile)
}
load(logrankfile)
head(log_rank_p)
log_rank <- as.data.frame(log_rank_p) %>%
rownames_to_column(var = "gene") %>%
arrange(log_rank_p)
table(log_rank_p < 0.01)
table(log_rank_p < 0.05) 11.4.4 批量单因素 cox
coxfile <- paste0(proj,"_cox.Rdata")
if(!file.exists(coxfile)){
cox_results <-apply(exprSet , 1 , genecox)
cox_results=as.data.frame(t(cox_results))
save(cox_results,file = coxfile)
}
load(coxfile)
table(cox_results$p<0.01)
table(cox_results$p<0.05)
lr = names(log_rank_p)[log_rank_p<0.01];length(lr)
cox = rownames(cox_results)[cox_results$p<0.01];length(cox)
length(intersect(lr,cox))
# save(lr,cox,file = paste0(proj,"_logrank_cox_gene.Rdata"))